Tin xem nhiều






Dự đoán về cơ chế và tính chất nhiệt hóa của phản ứng khí quyển O 3 + H 2 S
Cơ chế phản ứng ôzôn và hydro sunfua bao gồm một phức hợp đã được nghiên cứu ở B3LYP / 6-311 ++ G (3df, 3pd) và CCSD / 6-311 ++ G (3df, 3pd) // B3LYP / 6-311 ++ G (3df, 3pd) mức độ tính toán. Sự tương tác giữa nguyên tử lưu huỳnh của hydro sunfua và nguyên tử oxy của ozon tạo ra phức chất H2S-O3 bền vững. Với sự phân hủy của phức hợp này, bốn sản phẩm đã được tìm thấy. Các phân tích tôpô của nguyên tử trong phân tử và tính toán tần số dao động đã được sử dụng để xác nhận cơ chế. Chỉ số nhiệt động lực học tại T = 298,15 K và áp suất khí quyển đã được tính toán. Kết quả cho thấy việc tạo H2O + SO2 là kênh phản ứng chính với Δ G ° = −645,84 kJ / mol. Hằng số tốc độ của phản ứng H2S + O3 cho thấy hai kênh sản phẩm, SO2 + H2O và HSO + HOO, cạnh tranh với nhau dựa trên nhiệt độ.
1. Giới thiệu
Ozone và hydro sunfua là hai chất gây ô nhiễm khí quyển nghiêm trọng. Ozone đóng một vai trò quan trọng trong tầng cao của bầu khí quyển. Nồng độ ôzôn tương đối lớn trong khí quyển. Gần đây, phản ứng của ozon với các chất khác trong khí quyển đã được nghiên cứu nhiều trong lý thuyết và thực nghiệm.
Các hợp chất lưu huỳnh được thải vào khí quyển dưới nhiều dạng khác nhau thông qua các nguồn sinh học (thối rữa sinh khối và phun trào núi lửa) và do con người (công nghiệp hóa chất). Hydro sunfua có độc tính cao và được sử dụng trong một số ngành công nghiệp hóa chất.
Phản ứng của hydro sunfua với các phân tử và gốc khác nhau trong tầng bình lưu là chủ đề của nhiều nghiên cứu lý thuyết và thực nghiệm. Trong các nghiên cứu thực nghiệm, H2S phản ứng với ozon để tạo ra SO2 và H2O, là một trong những nguồn gây ra mưa axit.
Phản ứng của ozon và H2S có thể hỗ trợ loại bỏ chất ô nhiễm độc hại quan trọng trong khí quyển. Hơn nữa, một phản ứng pha khí chậm của ozon với hydro sunfua có thể là đáng kể nếu phản ứng tiếp tục tạo ra (HSOH) và (HSOOOH) - nguồn quan trọng của các gốc OH, HS và HOO (hydroperoxyl) vốn được biết đến nhiều như chất oxy hóa và tham gia vào quá trình phá hủy ozon. Do đó, trong nghiên cứu này, nhóm tác giả đã nghiên cứu tính toán chi tiết về cơ chế và nhiệt động lực học của phản ứng hydro sunfua và ozon. Ngoài ra, phân tử hydro thioperoxide được chỉ định là sản phẩm mới khác của các chất ban đầu.
Phương pháp
Dạng hình học của chất phản ứng, phức chất (ký hiệu là C), sản phẩm và trạng thái chuyển tiếp (ký hiệu là TS) tham gia vào phản ứng được tối ưu hóa bằng cách sử dụng lý thuyết hàm mật độ tại spin không hạn chế B3LYP với 6-311 ++ G (3df, 3pd ) bộ bazơ. Để có được mức năng lượng tương đối cao hơn, đáng tin cậy hơn của phương pháp tương quan điện tử, CCSD (T) / 6-311 ++ G (3df, 3pd) // B3LYP / 6-311 ++ G (3df, 3pd), được sử dụng trong đơn tính toán năng lượng điểm để cải thiện độ chính xác của thông tin năng lượng trên đường năng lượng tối thiểu. Năng lượng phân tử ở mức CCSD (T) và năng lượng điểm 0 (ZPE) thu được từ mức B3LYP được báo cáo. Tần số dao động điều hòa thu được ở mức B3LYP để xác minh các hình học được tối ưu hóa. Bản chất của các điểm dừng và trạng thái chuyển tiếp được xác định theo số giá trị riêng âm của ma trận Hessian. Ngoài ra, tọa độ phản ứng nội tại (IRC) được thực hiện để kiểm tra mối liên hệ giữa tất cả các loại tham gia phản ứng. Quy trình đối ứng được sử dụng để hiệu chỉnh năng lượng tương tác đối với lỗi tập cơ sở và phương pháp CCSD (T) được sử dụng để tính toán giá trị chẩn đoán cho các cấu trúc.
Ở cấp độ của phương pháp này, có thể tạo ra một hàm sóng ở dạng thích hợp để thực hiện phân tích cấu trúc liên kết các nguyên tử trong phân tử bằng cách sử dụng các chương trình dòng AIM2000. Hơn nữa, dữ liệu nhiệt động lực học đã được tính toán bằng cách sử dụng cơ học thống kê. Hằng số tốc độ của các kênh phản ứng cho các sản phẩm nhiệt động và động học đã được tính toán bằng lý thuyết TST và RRKM nhờ phương pháp B3LYP / 6-311 ++ G (3df, 3pd). Tất cả các tính toán được báo cáo trong công trình này được thực hiện với gói chương trình Gaussian 03.
2. Kết quả và Thảo luận
Hình dạng tối ưu hóa của chất phản ứng, phức chất (ký hiệu là C ), trạng thái chuyển tiếp (ký hiệu là TS), và các sản phẩm với dữ liệu thực nghiệm có sẵn tương ứng cho một số loại được thể hiện trong Hình 1. Kết quả tối ưu hóa hình học cho tất cả các loài thu được ở mức lý thuyết B3LYP / 6-311 ++ G (3df, 3pd) trong công trình này phù hợp với các giá trị thực nghiệm.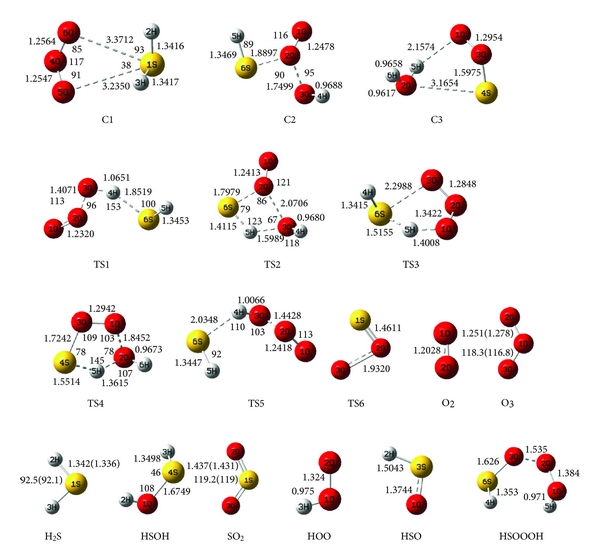
Một phức chất phản ứng, H2S-O3 (ký hiệu là C1), đã được hình thành giữa các chất phản ứng trên bề mặt thế năng đơn. Trong phức chất, các nguyên tử oxy của O 3 liên kết với nguyên tử lưu huỳnh của H2S và một vòng bốn thành viên được hình thành. Hình học phân tử chỉ ra rằng phức chất C1 có cấu trúc không đối xứng.
Trong phức chất C1, độ dài liên kết của S – O1 và S – O2 mới được tạo thành tương ứng là 3,371 và 3,235 Å ở mức B3LYP, và liên kết của O – O dài hơn khoảng 0,004 so với liên kết tương ứng ở cấp độ gốc phân tử ozon. Để xác định sự hình thành liên kết mới và sự tồn tại của cấu trúc vòng, nhóm nghiên cứu đã sử dụng phân tích tôpô về mật độ điện tích. Giá trị của các tham số điểm tới hạn của liên kết và gradien mật độ điện tử đối với các liên kết mới hình thành và điểm tới hạn của vòng đối với cấu trúc vòng được tính toán trong Hình 2. Giá trị mật độ điện tích điện tử Laplacian của liên kết S – O1 và S – O2 lần lượt là 0,0262 và 0,0265 au. Cả giá trị thấp và dương của Laplacian từ phân tích AIM đối với liên kết S – O của phức chất C1 cho thấy rằng có một tương tác van der Waals yếu giữa nguyên tử lưu huỳnh của H2S và nguyên tử oxy đầu cuối của ozon. Mật độ điện tích tương ứng với các liên kết mới lần lượt là 0,0095 và 0,0685 au. Mật độ điện tích được tính toán xác nhận tương tác yếu của ozon và hydro sunfua trong phức chất C1.
Năng lượng liên kết của C1 thấp hơn 5,78 kJ / mol so với các chất phản ứng ban đầu (H2S + O3 ) ở mức B3LYP. Giá trị sai số chồng chất bộ cơ sở (BSSE) cho năng lượng phức chất C1 là 0,01 kJ / mol. Không có trạng thái chuyển tiếp nào được tìm thấy cho sự hình thành của nó. Vì vậy, sự hình thành của C1 là một quá trình không có rào cản. Sau đó, thông qua nhiều loại C1, ba loại sản phẩm thu được.
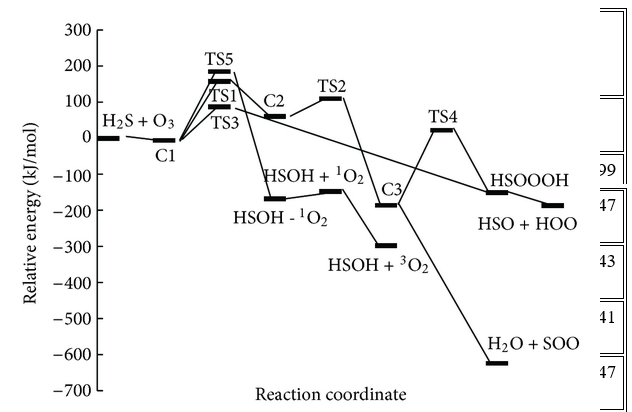
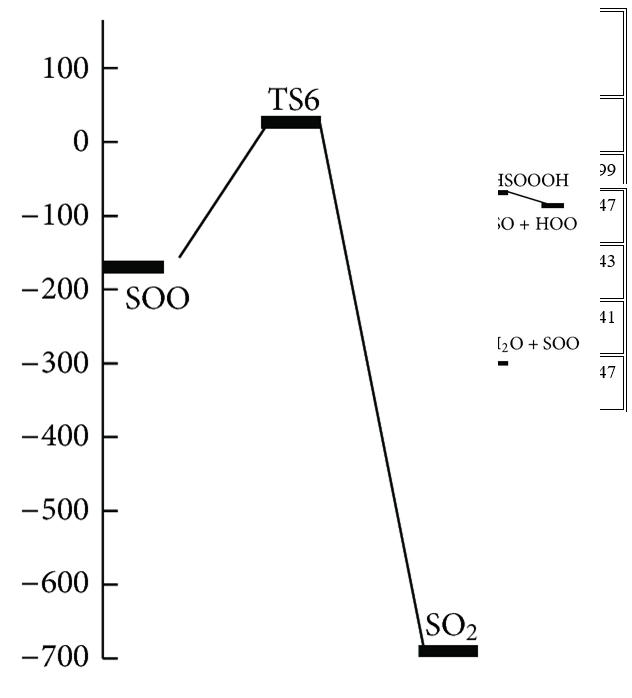
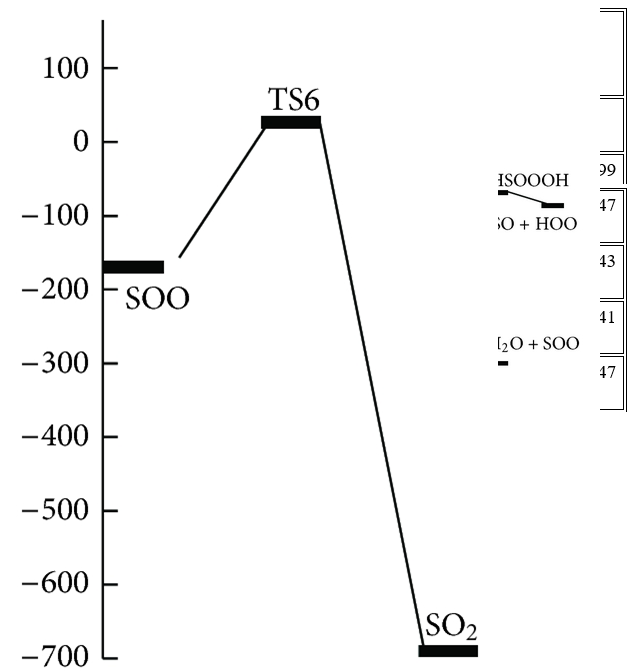
Trong những con đường này, tác giả đã tìm thấy năm trạng thái chuyển tiếp, TS1, TS2, TS3, TS4 và TS5, trên bề mặt thế năng của phản ứng H2S + O3 và một trạng thái khác, TS6, được mô tả là sự biến đổi của SOO thành SO2 và bốn điểm đứng yên khác (C2, C3, HSOOOH và SOO). Trên thực tế, đồ thị phân tử tương ứng (Hình 2) cho thấy sự tồn tại của BCP giữa các nguyên tử ngoại quan. Ngoài ra, cấu trúc vòng được hình thành khi các điểm tới hạn của vòng xuất hiện ở tất cả các loại. Tổng năng lượng và năng lượng tương đối của tất cả các loại ở B3LYP / 6-311 ++ G (3df, 3pd), dưới áp suất khí quyển và 298,15 K được liệt kê trong Bảng 1 và năng lượng tương đối CCSD (T) được thể hiện trong Hình 3 . Mức độ cao hơn của phương pháp tương quan điện tử CCSD (T) // B3LYP được sử dụng trong tính toán năng lượng điểm đơn lẻ để cải thiện độ chính xác của thông tin năng lượng trên đường năng lượng tối thiểu. Tần số dao động điều hòa thu được ở mức B3LYP để xác minh các hình học được tối ưu hóa. Ngoài ra, kết quả của các phép tính năng lượng điểm đơn lẻ ở mức tính toán CCSD / 6-311 ++ G (3df, 3pd) // B3LYP / 6-311 ++ G (3df, 3pd) được tóm tắt trong Bảng 1. Tất cả các năng lượng trong Bảng 1 được hiệu chỉnh bởi BSSE.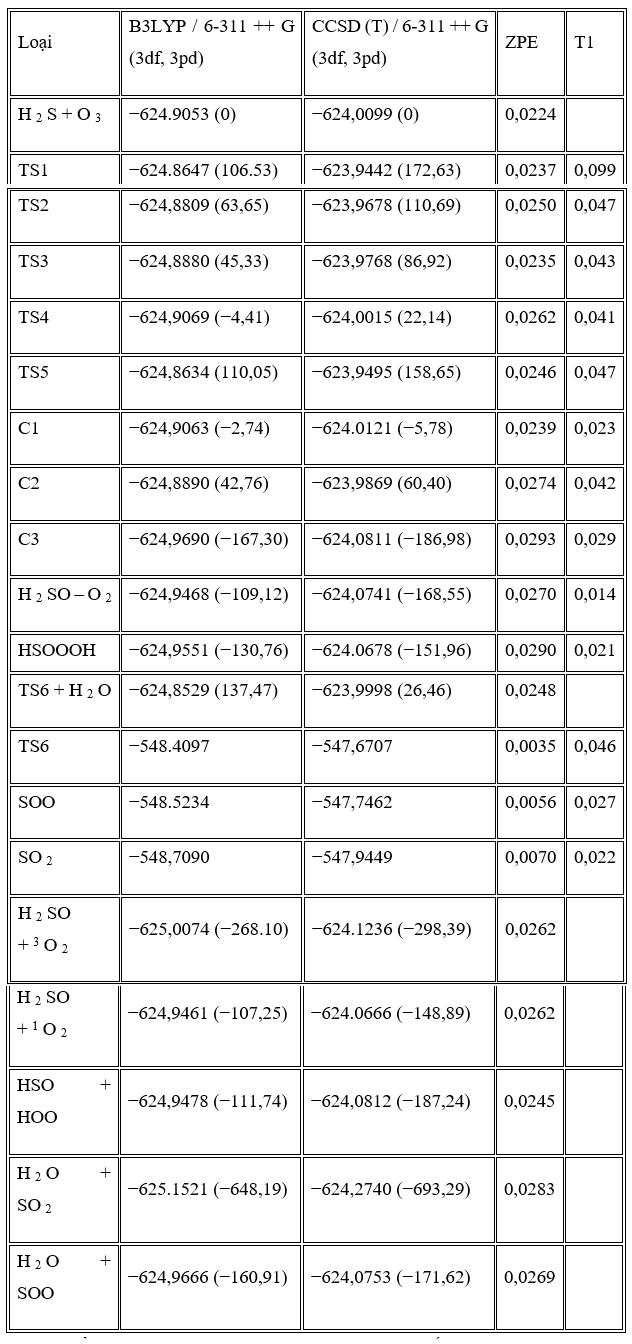
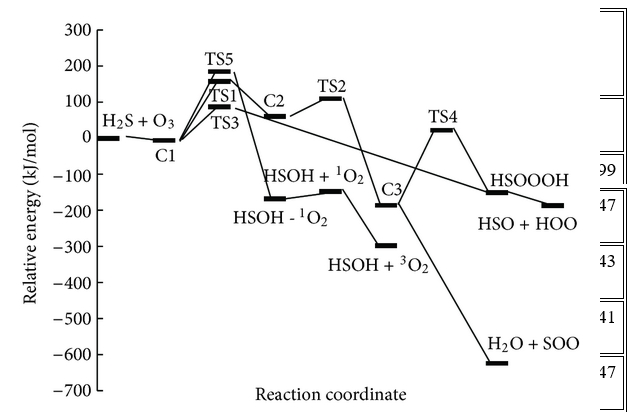
Các phản ứng trên các bề mặt thế năng đơn lẻ có thể được tóm tắt như sau:
P1 (1): O 3 + H 2 S → C1 → TS1 → C2 → TS2 → C3 → SOO + H 2 O → TS6 + H 2 O → SO 2 + H 2 OP1
(2): O 3 + H 2 S → C1 → TS1 → C2 → TS2 → C3 → TS4 → HSOOOH → HSO + HOOP2: O 3 + H 2 S → C1 → TS3 → HSOOOH → HSO + HOOP3: O 3 + H 2 S → C1 → TS5 → 1 O 2 + HSOH → 3 O 2 + HSOH
Trong đó HSOOOH là phức giữa các phân tử HOO và HSO. Các chi tiết của cơ chế phản ứng trên bề mặt thế năng singlet sẽ được thảo luận dưới đây.
2.1. Thuộc tính của đường phản ứng
Chúng ta chỉ có một phức chất keo (C1) nằm trên bề mặt năng lượng tiềm năng cho phản ứng H2S + O3, phản ứng này được hình thành khi các nguyên tử lưu huỳnh của hydro sunfua được gắn với các nguyên tử oxy cuối cùng của ozon. Đối với phản ứng H2S + O3 , có ba sản phẩm có thể có là SO2 + H2O, HSO + HOO và HSOH + O2 .
2.1.1. Kênh hình thành SO 2 + H 2 O
Trong con đường P1 (1), C1 trải qua quá trình hình thành liên kết H – O1 và S – O2 và quá trình phá vỡ liên kết S – O1, S – O3 và H – S để tạo thành C2 qua TS1 với rào cản năng lượng là 178,41 kJ / mol. Sau đó C2 chuyển thành C3 thông qua TS2 với rào cản năng lượng 50,29 kJ / mol bởi sự hình thành liên kết O1 – S và H – O3 và sự chuyển hydro từ nguyên tử lưu huỳnh sang nguyên tử O1.
Trong vùng chuyển tiếp cấu trúc vòng bốn phần tử (TS2) của quá trình C2 → C3, mật độ điện tích điện tử và Laplacian của nó lần lượt là 0,0356 và 0,1718 au.
Chiều cao năng lượng cho thấy chuyển đổi C1 → C3 là khả thi về mặt năng lượng với hai trạng thái chuyển đổi năng lượng thấp. Con đường phản ứng tiếp tục với sự hình thành các phân tử nước và SOO từ C3 mà không đi qua bất kỳ trạng thái chuyển tiếp nào. Cuối cùng, SOO trải qua quá trình hình thành liên kết O – S và quá trình đứt gãy liên kết O – O để hình thành phân tử SO2 thông qua TS6 với hàng rào năng lượng 26,46 kJ / mol so với chất phản ứng ban đầu và 198,08 kJ / mol so với phân tử SOO. Năng lượng của sản phẩm cuối cùng (H2O + SO2 ) thấp hơn 693,29 kJ / mol so với các chất phản ứng ban đầu (H2S + O3 ). Vì sự tạo thành SO2 có thể được tạo ra từ sự sắp xếp lại của SOO và số nguyên tử của chúng khác với những nguyên tử khác, nhóm nghiên cứu chỉ ra bước này của con đường P1 (1) riêng lẻ trong Hình 3 . Đồ thị phân tử tương ứng của tất cả các loài thu được từ các tính toán AIM trong đường dẫn P1 (1) trong Hình 2 xác nhận rằng sự hình thành và sự đứt gãy của các liên kết được chỉ ra bởi sự xuất hiện của BCP và sự sụp đổ của BCP tương ứng. Ngoài ra, tần số ảo của TS1, TS2 và TS6, biểu thị sự phân cắt của một số liên kết và sự hình thành các liên kết khác trong tọa độ phản ứng, lần lượt là 327i, 751i và 970i cm −1 . Trong tất cả các bước của đường P1 (1), sự hình thành các sản phẩm từ chất phản ứng được xác nhận bằng cách sử dụng các đường cong IRC liên quan.
2.1.2. Các kênh hình thành HSO + HOO
Đối với sản phẩm HSO + HOO, có hai con đường trên bề mặt năng lượng thế năng đơn ký hiệu là P1 (2) và P2 được xác nhận bằng cách sử dụng tần số dao động của các trạng thái chuyển tiếp, phân tích AIM và đường cong IRC cho các trạng thái chuyển tiếp.
Trong con đường P1 (2), sự hình thành của C3, cấu trúc vòng năm thành viên với giá trị thấp của mật độ điện tích electron, 0,0081 au, và Laplacian của mật độ điện tích, 0,0366 au, tương tự như con đường P1 (1). C3 trải qua quá trình đứt gãy liên kết H – O1 và biến mất cấu trúc vòng để tạo thành HSOOOH thông qua TS4 với hàng rào năng lượng là 209,12 kJ / mol. Cuối cùng, các phân tử HSO + HOO được hình thành bằng cách phá vỡ liên kết O2 – O3 mà không trải qua bất kỳ trạng thái chuyển tiếp nào. Sự hình thành các sản phẩm từ C3 được xác nhận bằng cách sử dụng kết quả AIM và đường cong IRC.
Trong con đường P2, C1 chuyển thành phức HSOOOH và các sản phẩm tương ứng của nó (HSO và OOH) trong phản ứng một bước thông qua TS3 với hàng rào năng lượng là 92,70 kJ / mol. Sự biến đổi của nó, đi kèm với sự hình thành liên kết H2 – O3 và quá trình đứt gãy liên kết S – O3 để tạo thành một trạng thái chuyển tiếp vòng năm nhớ, TS3, phân tử. Sau đó, cấu trúc vòng của TS3 biến mất và hình thành mẫu HSOOOH. Vòng tồn tại, liên kết H2-O3, sự hình thành và sự phân cắt liên kết S-O3 được xác nhận bằng cách sử dụng phép tính AIM được toán học trong Hình 2 .
2.1.3. Kênh hình thành HSOH + O2
Một bước đơn giản khác của phản ứng là con đường P3 là một phản ứng cơ bản. Trong con đường P3, C1 trải qua các sản phẩm (HSOH + O2) bằng cách sử dụng quá trình tách oxy của ozon bằng hydro sunfua và đồng thời hydro chuyển từ nguyên tử lưu huỳnh sang nguyên tử oxy mới thông qua TS5 với rào cản năng lượng là 164,43 kJ / mol và dao động tần số ảo 500i . Sự trừu tượng oxy và sự dịch chuyển hydro dẫn đến sự hình thành HSOH được xác nhận bằng cách sử dụng phép tính tọa độ phản ứng nội tại (IRC).
Tóm lại, phản ứng H2S + O3 có tính khả thi về mặt năng lượng đối với việc hình thành cả ba sản phẩm trong các con đường tương ứng mà tất cả các bước của phản ứng đều xác nhận bằng cách sử dụng các tính toán IRC và AIM.
2.2. Phân tích tôpô về mật độ điện tử của các phân tử HSOH, HSO và SOO
Phân tích tôpô AIM về mật độ điện tích đã được xây dựng bằng cách sử dụng gói chương trình AIM2000. Phân tích tôpô AIM đã thu được bằng cách sử dụng mật độ điện tích tích hợp trên các lưu vực nguyên tử (lên đến 0,001 e / mức bohr 3 ) cũng như về mật độ điện tử , mật độ Laplacian và độ elip liên kết , tại các điểm tới hạn của liên kết (BCP) trong đó và là các giá trị riêng của ma trận Hessian của mật độ electron BCP.
Theo phân tích tôpô về mật độ điện tích, trong lý thuyết về nguyên tử trong phân tử (AIM), mật độ điện tử và Laplacian của mật độ điện tử , được sử dụng để mô tả độ bền và đặc tính của liên kết, tương ứng. Laplacian là giá trị riêng của ma trận Hessian của mật độ điện tử. Nếu một điểm tới hạn có hai giá trị riêng âm và một dương, nó được gọi là (3, −1) hoặc điểm tới hạn của trái phiếu (BCP). Nếu một điểm tới hạn có hai giá trị dương và một âm thì nó được gọi là (3, + 1) hoặc điểm tới hạn vòng (RCP), cho biết rằng có một cấu trúc vòng. Theo lý thuyết của AIM, Laplacian của mật độ electron ( ) mô tả đặc tính của liên kết. Nói chung, khi 0 liên kết thuộc tương tác tĩnh điện.
Phân tích tôpô của mật độ điện tử được thực hiện đối với tất cả các liên kết trong các loại HSOH, HSO và SOO là các sản phẩm tương đối ổn định của phản ứng. Các đặc điểm cấu trúc liên kết trong BCP của liên kết S – O, O – H và H – S được lập bảng trong Bảng 3 . Ngoài ra, các giá trị trong Bảng 3 chỉ ra rằng tất cả các liên kết trong HSOH đều có đặc tính cộng hóa trị ( H – S> S – O tương ứng với mật độ điện tích lần lượt là 0,3781, 0,2225 và 0,1929 au. Trong gốc HSO, liên kết H – S có tính chất cộng hóa trị trong khi liên kết S – O có bản chất tương tác tĩnh điện với mật độ điện tích lần lượt là 0,2174 và 0,2686 au. Mật độ điện tử của liên kết HS của HSOH so với liên kết tương ứng trong liên kết HSO cho thấy liên kết HSOH mạnh hơn liên kết tương ứng của HSO, còn liên kết S – O trong HSO có mật độ điện tích cao hơn liên kết tương ứng trong phân tử HSOH tương tự như liên kết đôi lưu huỳnh và oxy. Ở các loài SOO, liên kết O – O có đặc tính cộng hóa trị trong khi liên kết S – O có đặc tính tĩnh điện với các giá trị mật độ điện tử tương ứng là 0,4369 và 0,2048 au. Liên kết S – O lạ trong phân tử SOO yếu hơn liên kết tương ứng trong phân tử HSOH và mạnh hơn liên kết tương ứng trong HSO. Bản chất của nó giống với phân tử HSO.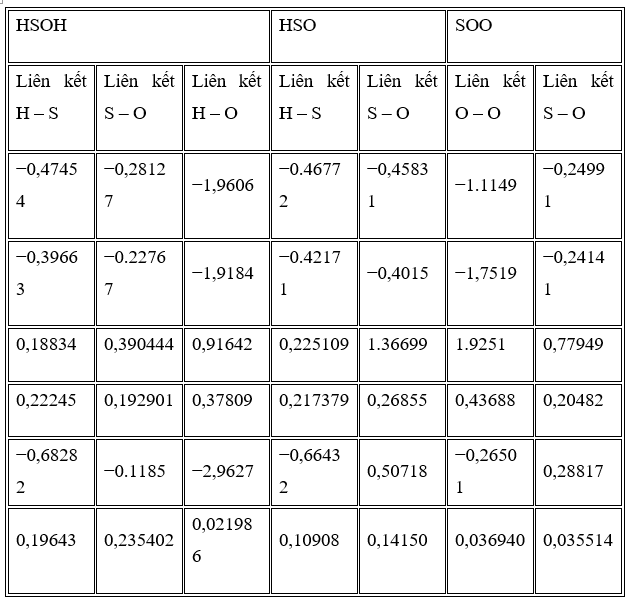
Độ elip là đại lượng đo tỷ số giữa tốc độ giảm mật độ theo hai phương vuông góc với đường liên kết tại điểm tới hạn của liên kết, hình dạng chung của liên kết và độ của ký tự π . Giá trị 0 biểu thị sự phân bố mật độ đối xứng về đường liên kết, chẳng hạn như phân bố mật độ được tìm thấy trong liên kết đơn và liên kết ba tiêu chuẩn, trong khi giá trị lớn biểu thị sự ưu tiên cho sự tích tụ mật độ theo một hướng cụ thể. Các giá trị về độ elip của các liên kết khác nhau đối với phân tử HSOH được liệt kê trong Bảng 3 . Tính elip cao ( ) đối với liên kết S – O trong cả phân tử HSOH và HSO cho thấy đặc tính của liên kết π cao , trong khi giá trị thấp thuộc về liên kết O – H và H – S tương ứng với liên kết π thấp. Trong phân tử SOO, cả hai liên kết đều có độ elip gần như nhau (0,0369 và 0,0352 au). Cách thức này cho thấy cả hai liên kết có sự đối xứng gần như giống nhau trong sự phân bố của đặc tính liên kết và sự phân bố của nó gần như đối xứng giữa hai liên kết. So sánh giữa hai liên kết cho thấy liên kết O – O có ký tự π cao hơn so với liên kết S – O, điều này được xác nhận bằng các dự đoán thực nghiệm.
2.3. Phân tích quỹ đạo phân tử
Ái lực điện tử, thế ion hóa, thế hóa học, độ cứng hóa học và một số thông số khác cho phản ứng hóa học được dự đoán bởi HOMO và phân tích các obitan LUMO. Năng lượng HOMO (obitan phân tử chiếm cao nhất) đặc trưng cho khả năng nhường electron; năng lượng LUMO (quỹ đạo phân tử không bị trống thấp nhất) đặc trưng cho khả năng nhận điện tử. Chúng ta có thể xác định cách phân tử tương tác với các chất khác; do đó, chúng được gọi là các obitan biên giới. Khoảng cách năng lượng giữa HOMO và LUMO đặc trưng cho việc các phân tử có ổn định về mặt hóa học hay không, và nó là một thông số quan trọng trong việc xác định các đặc tính vận chuyển điện phân tử vì nó là thước đo độ dẫn điện tử. Ngoài ra, năng lượng của HOMO liên quan trực tiếp đến thế ion hóa, trong khi lượng điện tử ái lực liên quan đến năng lượng của LUMO. Khoảng trống năng lượng chịu trách nhiệm phần lớn cho các đặc tính hóa học và quang phổ của các phân tử.
Các tính năng của HOMO và LUMO được tính bằng mức B3LYP / 6-311 ++ G (3df, 3pd) trong pha khí. Khoảng cách vùng năng lượng ( ) (chuyển từ HOMO sang LUMO), thế hóa học ( μ ), độ cứng hóa học ( ), thế ion hóa và ái lực điện tử của các loại HSOH, HSO và SOO có thể được trình bày trong Bảng 4 .
Bảng 4: Độ cứng ( η ), thế năng hóa học ( μ ), năng lượng HOMO ( ) và năng lượng LUMO ( ) (tính theo đơn vị nguyên tử) của phân tử HSOH và HSO.
Khoảng trống năng lượng HOMO-LUMO trong Bảng 4 dự đoán HSO (0,2358 au) với khoảng cách năng lượng lớn, ổn định hơn so với các phân tử HSOH (0,2052 au) và SOO (0,0937 au).
Các obitan phân tử biên của HSO và HSOH được mô tả trong Hình 4. Trong phân tử HSO, LUMO được định vị trên liên kết giữa các nguyên tử oxy-lưu huỳnh. HOMO chủ yếu được cấu tạo trên các obitan nguyên tử loại p của nguyên tử oxy và lưu huỳnh. Trong phân tử SOO, obitan HOMO nằm trên nguyên tử lưu huỳnh trong khi LUMO được cấu tạo trên obitan loại π của tất cả các nguyên tử.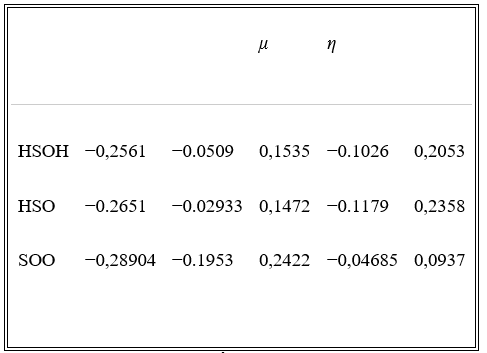
Khoảng trống năng lượng HOMO-LUMO trong Bảng 4 dự đoán HSO (0,2358 au) với khoảng cách năng lượng lớn ( ) ổn định hơn so với các phân tử HSOH (0,2052 au) và SOO (0,0937 au).
Các obitan phân tử biên của HSO và HSOH được mô tả trong Hình 4 . Trong phân tử HSO, LUMO được định vị trên liên kết giữa các nguyên tử oxy-lưu huỳnh. HOMO chủ yếu được cấu tạo trên các obitan nguyên tử loại p của nguyên tử oxy và lưu huỳnh. Trong phân tử SOO, obitan HOMO nằm trên nguyên tử lưu huỳnh trong khi LUMO được cấu tạo trên obitan loại π của tất cả các nguyên tử.
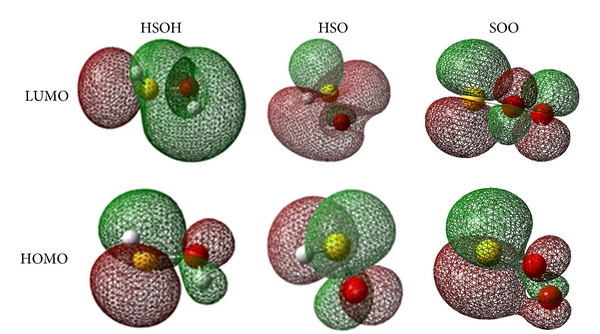
Hình 4: Các obitan phân tử biên của phân tử HSO, HSOH và SOO.
Điện thế hóa học ( μ ) và độ cứng hóa học ( ) đã được xác định bằng cách sử dụng năng lượng HOMO và LUMO được tính toán bằng cách sử dụng các quan hệ và đâu là thế ion hóa ( ) và là ái lực điện tử ( ) của hệ phân tử. Theo nguyên tắc độ cứng tối đa, các anion ổn định nhất phải có giá trị độ cứng lớn nhất là một cấu trúc năng lượng tối thiểu ở một thế hóa học không đổi. Dữ liệu trong bảng 4 cho thấy rằng giá trị tuyệt đối lớn nhất của độ cứng hóa học, −0,179 au, thuộc về phân tử HSO. Ngoài ra, thế hóa học tối thiểu, 0,1472 au, của phân tử HSO phù hợp với thế năng hóa học dự đoán.
Xác định sự phân bố điện tích phân tử là cơ sở cho việc áp dụng phương pháp mô hình phân tử vào một hệ thống hóa học thực tế. Sự thay đổi trong phân bố điện tích phân tử với cấu trúc có thể được áp dụng để giải thích cho sự ổn định của các loài. Các điện tích nguyên tử được tìm thấy bên trong các nguyên tử được thể hiện trong Hình 5 . Tất cả các điện tích nguyên tử của các phân tử HSOH, HSO và SOO đều thu được bằng phương pháp Mulliken ở mức B3LYP / 6-311 ++ G (3df, 3pd). Kết quả cho thấy các nguyên tử lưu huỳnh mang điện tích dương trong ba loại và các nguyên tử oxy mang điện tích âm trong cả hai phân tử HSOH và HSO, và trong SOO, nguyên tử oxy đầu cuối mang điện tích âm trong khi các nguyên tử khác mang điện tích dương như trong Hình 5. Nhưng giá trị tuyệt đối của sự phân bố điện tích của nguyên tử oxy trong SOO nhỏ hơn phân tử HSO và điện tích âm oxy của nó nhỏ hơn phân tử HSOH tương ứng với thế hóa học và phân tích quỹ đạo phân tử HOMO-LUMO.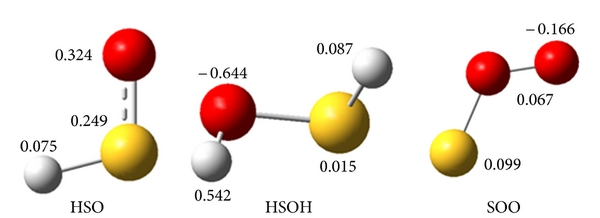
Mômen lưỡng cực cho thấy sự phân bố điện tích phân tử và được cho dưới dạng véc tơ trong không gian ba chiều. Do đó, nó có thể được sử dụng như một bộ mô tả để mô tả sự chuyển động của điện tích trong phân tử. Hướng của vectơ mômen lưỡng cực trong phân tử phụ thuộc vào tâm của các điện tích dương và âm. Ở các loài HSO, vectơ mômen lưỡng cực (2,298 D) là từ nguyên tử lưu huỳnh đến nguyên tử oxi song song với độ dài liên kết S – O nhưng trong phân tử HSOH vectơ này (1,594 D) nằm ở bên ngoài phân tử và có thể gây ra độ ổn định thấp trong phân tử. Đối với phân tử SOO, vectơ này (1,525 D) là từ nguyên tử oxy đầu cuối đến nguyên tử lưu huỳnh.
Hằng số quay của phân tử HSOH là 202,1, 15,2 và 14,7 GHz và đối với HSO là 298,3, 20,3 và 19,0 GHz và đối với các loài SOO là 95,2, 6,9 và 6,4 GHz. Bởi vì giá trị thực nghiệm của các hằng số quay không thể truy cập được, vì vậy kết quả tính toán được báo cáo ở dạng tuyệt đối.
2.4. Dữ liệu nhiệt động lực học trong quá trình phản ứng O3 và H2S
Sự thay đổi của các đặc tính nhiệt động đối với mỗi kênh phản ứng là sự khác biệt giữa các đặc tính nhiệt động tương ứng của sản phẩm và chất phản ứng. Dữ liệu nhiệt động lực học được tính toán bằng mức tính toán G4 trong pha khí và được ZPE hiệu chỉnh cho phản ứng O3 + H2S.
Năng lượng nội tại tương đối tính được, entanpi, năng lượng tự do Gibbs và entropi của tất cả các sản phẩm của các phản ứng trong pha khí ở áp suất khí quyển và nhiệt độ 298,15 K được tóm tắt trong Bảng 5 .
Phản ứng 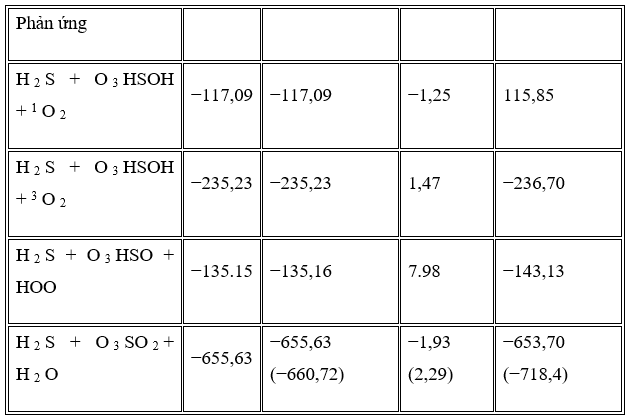
Dữ liệu bảng 5 cho thấy rằng Δ của tất cả các kênh sản phẩm tổng thể đều âm có nghĩa là các đường phản ứng tỏa nhiệt. Giá trị này đối với đường dẫn SO2 + H2O lớn hơn các đường khác có nhiệt lượng dư cao. Ngoài ra, giá trị năng lượng tự do Gibbs của tất cả các kênh sản xuất đều âm nên tất cả các kênh sản phẩm đều tự phát trong pha khí ở áp suất khí quyển và 298,15 K. Kênh sản phẩm đường dẫn SO2 + H2O về mặt nhiệt động là khả thi nhất so với các sản phẩm khác bằng −653,70 kJ / mol ở năng lượng tự do Gibbs. Dữ liệu tính toán được so sánh với giá trị thực nghiệm của SO 2 + H 2. Kênh phản ứng O và các kết quả thu được có thể chấp nhận được so với thí nghiệm (sai số tương đối 9% trong năng lượng tự do Gibbs).
2.5. Tính toán các hằng số tỷ giá
Hằng số tốc độ là một tham số quan trọng trong nghiên cứu động học của phản ứng có thể được tính toán từ cơ học thống kê và phương trình của nó thu được từ lý thuyết trạng thái chuyển tiếp (TST) dựa trên cơ học thống kê. Đối với phản ứng hai phân tử, người ta thường sử dụng lý thuyết trạng thái chuyển tiếp, lý thuyết này đòi hỏi kiến thức về cấu trúc chuyển tiếp chính xác để năng lượng hoạt hóa được biết đến. Sau đây là một phương trình quen thuộc để tính hằng số tốc độ cho phản ứng hai phân tử, ban đầu xuất phát từ Eyring.
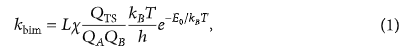
L là hằng số Avogadro; là rào cản năng lượng giữa TS và chất phản ứng bao gồm hiệu chỉnh năng lượng điểm 0; , và lần lượt là các chức năng phân vùng của cấu trúc chuyển tiếp và các loại chất phản ứng. Các tham số cho một loài là tích số của các hàm tịnh tiến, quay, dao động và phân vùng điện tử, là hằng số Boltzmann và là hằng số Planck.
Hàm phân hoạch tịnh tiến trên một đơn vị thể tích của phân tử khối trong không gian ba chiều là
Hàm phân vùng quay ngoài cho phân tử phi tuyến có dạng sau:
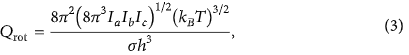
Các I tham số là mômen quán tính và là một số đối xứng. Khi hai chất phản ứng tiến lại gần nhau, ba chuyển động quay bên ngoài (đối với phân tử phi tuyến) hoặc hai (đối với phân tử tuyến tính) sẽ biến đổi thành chuyển động quay bị cản trở hoặc dao động điều hòa khi chúng tiếp cận trạng thái chuyển tiếp.
Rung động ở trạng thái chuyển tiếp thường được coi là dao động điều hòa. Hàm phân vùng dao động cho một bậc tự do được tính như
v là tần số dao động. Theo cơ học cổ điển, một hạt chỉ có thể vượt qua rào cản năng lượng tiềm năng nếu năng lượng của nó cao hơn độ cao của rào cản, trong khi cơ học lượng tử dự đoán rằng có xác suất vượt qua ở năng lượng hạt thấp hơn. Xác suất truyền này rất nhạy cảm với năng lượng và khối lượng của hạt, chiều cao của rào cản và hình dạng của rào cản. Hệ số hiệu chỉnh đường hầm được định nghĩa là thương số của tốc độ cơ lượng tử so với tốc độ cổ điển. Mặc dù quá trình đào hầm là một hiện tượng đa chiều, nhưng để đơn giản đây được coi là quá trình một chiều. Một nguyên nhân có thể gây ra sự sai lệch so với hành vi Arrhenius là sự đào hầm cơ học lượng tử của các chất phản ứng qua hàng rào cổ điển được giả định là bảo toàn tần số dao động và mômen quán tính của các mảnh trong quá trình phản ứng. Trong nghiên cứu này, chúng tôi đã sử dụng cách tiếp cận đơn giản nhất để đánh giá vai trò của đường hầm lượng tử là hiệu chỉnh Wigner đối với tốc độ phản ứng. Nó liên quan đến độ lớn của tần số ảo dọc theo tọa độ phản ứng ở trạng thái chuyển tiếp. Hằng số tốc độ được tăng cường bởi một hệ số
V(im):
Trong đó là tần số ảo có giá trị âm tính cho chuyển động dao động dọc theo đường phản ứng và là tốc độ ánh sáng.V(im)C
Hằng số tốc độ cho kênh phản ứng đơn phân tử được tính bằng lý thuyết RRKM (Rice-Ramsperger-Kassel-Marcus). Trong lý thuyết RRKM, hệ số tốc độ nhiệt nhận được bằng cách tích phân từ đến vô cùng của phương trình sau:
Và được xác định dựa trên cơ chế Lindemann cho các phản ứng hóa bậc nhất. Các thuật ngữ khác nhau của biểu thức tỷ lệ hiện được đánh giá bằng cách sử dụng cơ học thống kê. Trong giới hạn áp suất cao, lý thuyết RRKM giảm thành lý thuyết trạng thái chuyển tiếp và kết quả của hai lý thuyết trùng khớp với nhau.
Hằng số tốc độ của phản ứng đã được tính toán trên các bề mặt thế năng đơn lẻ cho hai con đường được xác định là đường nhiệt động (P1 (1)) và đường động học (P2) bằng RRKM (phản ứng đa kênh) và TST (phản ứng một bước) được thực hiện trong các chương trình Ssumes và Gpop.
Đối với phản ứng đa kênh (đường P1 (1)), hằng số tốc độ tổng được đánh giá trong giới hạn áp suất cao của cơ chế Lindemann. Trong giới hạn áp suất cao, lý thuyết RRKM giảm thành lý thuyết trạng thái chuyển tiếp và kết quả của hai lý thuyết trùng khớp với nhau. Giới hạn áp suất cao của hằng số tốc độ tổng được xác định dựa trên cơ chế Lindemann, là biểu thức tốc độ cho các bước thuận của phản ứng và và là các bước ngược lại của phản ứng theo phản ứng đơn phân tử. Đối với các kênh có một trạng thái chuyển tiếp, P2, hằng số tốc độ tổng được tính toán bằng cách sử dụng lý thuyết trạng thái chuyển tiếp cho các phản ứng hai phân tử (phương trình Eyring (1)).
Trên bề mặt thế năng đơn lẻ, bốn đường phản ứng, P1 (1), P1 (2), P2 và P3, thu được. Các hằng số tốc độ đã được tính toán cho hai đường (P1 (1) là đường nhiệt động và P2 là đường động học). Con đường đầu tiên, P1 (1), có sự kết hợp của hai bước, do đó, nó là một phản ứng phức tạp. Một là sự hình thành SOO và H 2 O dưới dạng sản phẩm và, phân tử SOO tiếp theo được sắp xếp lại thành phân tử SO 2 như một sản phẩm cuối cùng sau khi đi qua TS6. Trong phép tính hằng số tốc độ, bước cuối cùng (SOO → SO 2) đã bị bỏ qua vì nó là một phản ứng đơn phân tử. Vì vậy, nó nhanh hơn sự hình thành các bước SOO. Ngoài ra, số lượng nguyên tử ở trạng thái chuyển tiếp khác với số lượng nguyên tử trong các TS khác, đó là một hạn chế đối với phần mềm. Các sản phẩm phụ HSO và HOO sản xuất bằng hai kênh. Kênh đầu tiên để sản xuất các phân tử HSO và HOO là đi qua TS3 và kênh thứ hai là đi qua TS1, TS2 và TS4. Giữa chúng, cơ chế bao gồm TS3 đáng tin cậy hơn kênh ba bước khác và hằng số tốc độ được tính cho đường dẫn đáng tin cậy (P2).
Đối với phản ứng đa kênh của H 2 S và O 3 , hằng số tốc độ tính toán cho hai đường được ký hiệu là tương ứng với đường dẫn P1 (1) và P2, tương ứng. Bảng 6 cho thấy hằng số tốc độ riêng của hai đường dẫn ở khoảng nhiệt độ 200–2500 K ở mức B3LYP / 6-311 ++ G (3df, 3pd). Các giá trị hằng số tốc độ cho các đường dẫn trong Bảng 6 cho thấy rằng đường dẫn P2 về mặt động học là đường dẫn chính trong tất cả các dải nhiệt độ. Do đó việc tạo ra các phân tử HSO và HOO là sản phẩm chính. Do năng lượng mức thấp của trạng thái chuyển tiếp từ kênh TS3, sản xuất HSO + HOO là các sản phẩm phụ quan trọng nhất (đường dẫn P2 và tương ứng) so với các đường dẫn khác. Giá trị của hằng số tốc độ cho thấy sự cạnh tranh giữa đường dẫn P1 (1) và P2, đường dẫn P2 đáng tin cậy hơn đường dẫn P1 (1). Nói cách khác, phản ứng động học H 2 S + O 3 tiến hành HSO + HOO là sản phẩm chính trên bề mặt thế năng singlet thông qua con đường P2 thông qua các hằng số tốc độ của . Theo khía cạnh nhiệt động lực học, các chất cộng H 2 O + SO 2 là sản phẩm chính trên bề mặt thế năng đơn thông qua đường P1 (1) thông qua các hằng số tốc độ của . Hình 6 cho thấy các đồ thị Arrhenius cho các hằng số tốc độ và tỷ lệ.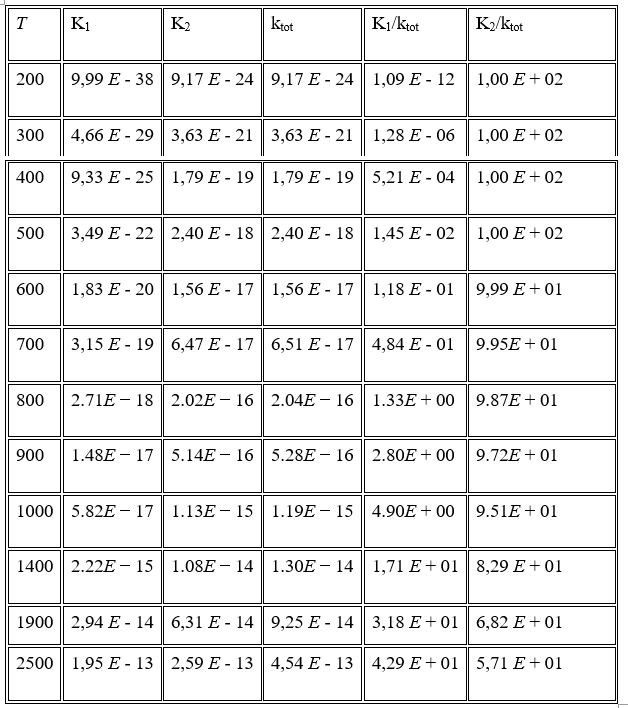
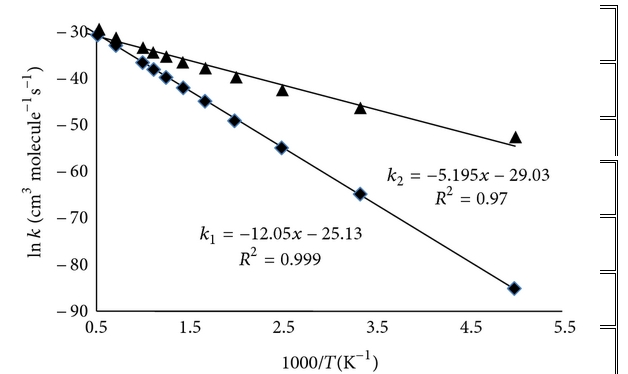 Hình 6: Biểu đồ Arrhenius cho hằng số tốc độ (- ∎ -) và (- ▲ -) của phản ứng H 2 S + O 3 .
Hình 6: Biểu đồ Arrhenius cho hằng số tốc độ (- ∎ -) và (- ▲ -) của phản ứng H 2 S + O 3 .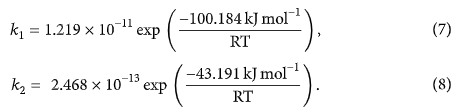
Như thể hiện trong Bảng 6, các giá trị hằng số tốc độ của H2S + O3 dưới áp suất khí quyển tăng khi nhiệt độ tăng. Do đó, sản phẩm chính của phản ứng phụ thuộc vào nhiệt độ và cơ chế phản ứng, do đó, phản ứng này có thể đóng những vai trò khác nhau trong việc hình thành mưa axit.
3. Kết luận
Bài viết khác

Sự nóng lên của Trái đất đang thay đổi với tốc độ chóng mặt, ngoài những điều dự đoán của con người. Kể từ giữa những năm 1800, khi con người bắt đầu đốt nhiên liệu hóa thạch ở quy mô công nghiệp, chúng ta đã tạo ra các chất đặc biệt gây hại cho Trái đất. Điều này được thể hiện ở nhiều yếu tố khác nhau

Nguyên tố đất hiếm ngày càng thu hút được nhiều sự quan tâm của giới khoa học và công nghiệp vì đặc tính và vai trò chủ chốt của chúng trong một số lĩnh vực nhưng sự lắng đọng của các nguyên tố đất hiếm tự nhiên còn hạn chế, dẫn đến sự phụ thuộc nhiều vào các nguồn tài nguyên thứ cấp. Than hoạt tính là phương pháp thu hồi đất hiếm đạt hiệu quả cao.

Phát triển kinh tế gắn với bảo vệ môi trường trong sản xuất kinh doanh, dịch vụ là xu thế tất yếu và nhu cầu cấp thiết trong tiến trình phát triển của mọi quốc gia, trong đó có Việt Nam. Sản xuất, kinh doanh phải bảo vệ môi trường là hình thức phát triển kinh tế bền vững theo hướng công nghiệp hóa hiện đại hóa, bằng cách sử dụng nguồn tài nguyên hợp lý, tiết kiệm và phòng ngừa, ngăn chặn, xử lý ô nhiễm môi trường hiệu quả đồng thời phát triển tài nguyên tái sinh.

Khử mùi ô tô là một trong những vấn đề nan giải của những quý ông khi sở hữu 1 chiếc xế. Nguyên nhân gây ra mùi do đâu, những nguyên nhân mà bạn đọc được ở dưới đây sẽ cho bạn biết vì sao xe bạn có mùi!

Khói thuốc lá có nhiều ảnh hưởng tới sức khỏe của con người, theo những nghiên cứu gần đây của các giáo sư đại học tại Mỹ thì khói thuốc lá còn làm cho người hút và người hít phải khói thuốc lá bị tăng cân dẫn đến béo phì - một căn bệnh cũng được coi là nguy hiểm trên thế giới

Tia UV-C được chứng minh là có khả năng làm bất hoạt Virus COVID-19. Kết quả này mở ra hướng mới cho nhân loại trong việc ngăn chặn, giảm thiểu các hệ quả và ngăn ngừa loại virus mới này.

Sử dụng máy lọc không khí máy khử mùi, máy lọc không khí ngày càng phổ biến. Đặc biệt là tại các vùng đất có mức độ ô nhiễm cao nhất trên thế giới sau đây

Ô nhiễm không khí bị ảnh hưởng ...



